
Statistical genetics for all occasions
Ever since I left my academic position and became independent, I have been interested in the idea of using minimal computer resources to perform big-data statistical analyses. I do not have a permanent office, so the only computer I own is a laptop, although it is a close to maximal-spec 15-inch MacBook Pro (mid-2015). I can use Amazon’s AWS for really big jobs, but the necessity to quickly trouble-shoot software and pipelines remains.
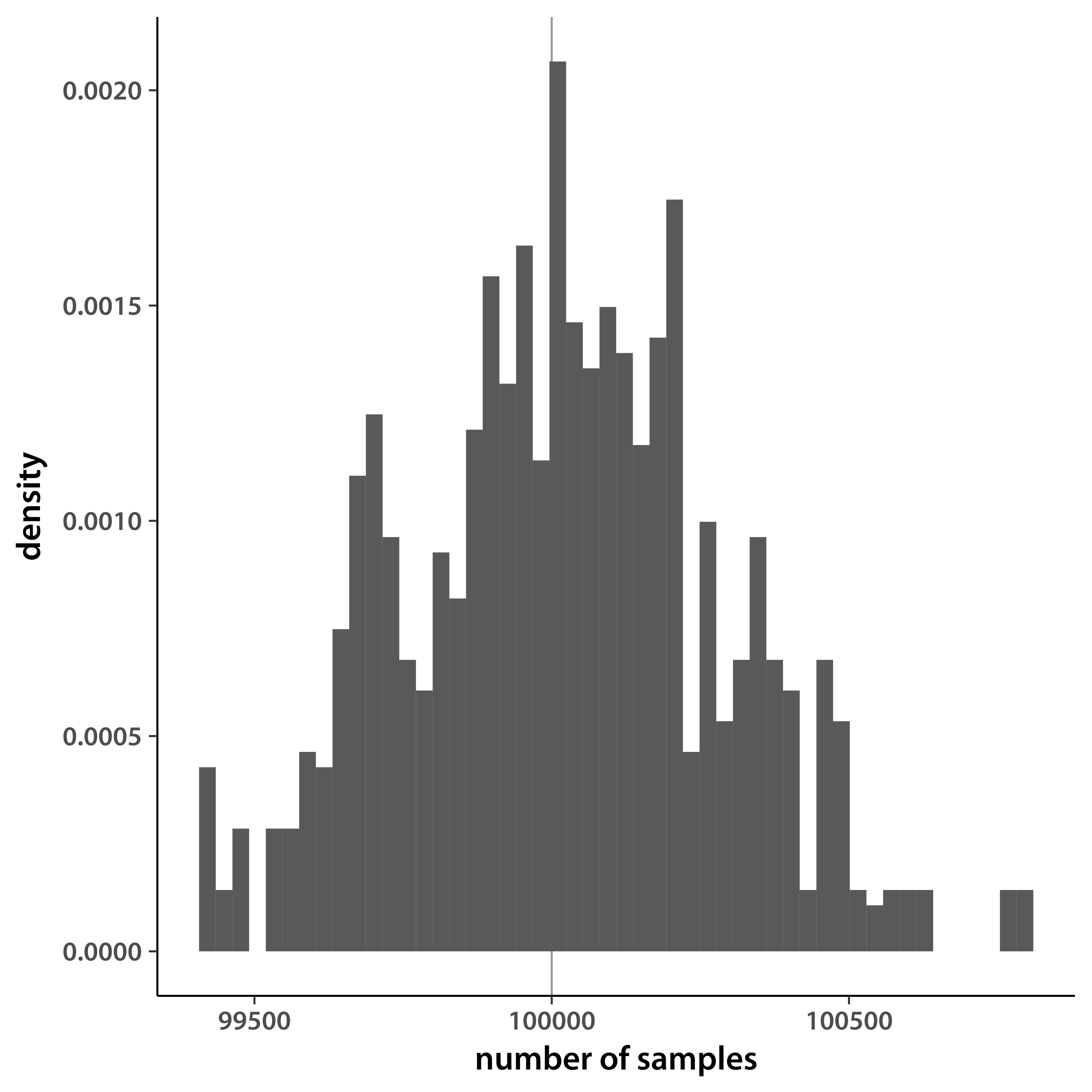
The major bottleneck for me is the size of DNA variant data sets. Phenotype data are increasing as well, but so far lag genomic sets by a couple of orders of magnitude. Binary compression, most notably the .bed format developed by the plink team, goes a long way towards making it possible to manipulate large genotype data sets on a laptop. To speed development even further, I needed to generate random subsets of large variant files. The samples had to reflect whole-data-set properties, most importantly loci had to be ordered along chromosomes the same as in the original file. I wrote a tool I call sampleSNPs to do this. As I was using it, I realized that it can have many more applications. For example, samples could be used for jack-knife uncertainty estimates for whole-genome statistics. While I personally stick with .bed files, I extended the functionality to other popular variant file formats (VCF, TPED, and HapMap). The only program I could find that also does this is plink, but it does it wrong. You give plink the fraction of variants you need (say, 0.2) and it will include each locus with that probability. As a result, the total number of SNPs in your sample varies from run to run. Here is an illustration, after running plink 1000 times:
When helping people with genome-wide association, mapping, and genome prediction analyses I am frequently asked to estimate the rate of decline of linkage disequilibrium with distance. This problem seems easy, but it blows up quickly as the number of marker grows and it becomes infeasible to estimate all pairwise LD. Sliding window approaches work, but waste disk space by over-sampling loci that are close to each other. I applied my sampling scheme to pairs of markers, resulting in a quick method that still produces reasonable estimates while using trivial computer resources.
The sampling scheme itself deserves a mention. Designing an algorithm like this is surprisingly hard. While most classical computational problems of this sort have been solved in the late 70s and early 80s, the on-line ordered sampling algorithm solution did not appear until 1987 in a paper by Jeffrey Vitter. Maybe because it came later than most, this work has been languishing in relative obscurity. I stumbled upon it by chance while reading some unrelated blog posts on computational methods.
I came up with my own C++ implementation of the Vitter algorithm, using Kevin Lawler’s C code as a guide. I wrote up a manuscript, available as a pre-print here and here while I am working on getting it published in a journal. The whole project is on GitHub. The included C++ library I wrote as a basis for the stand-alone programs has additional functionality and can be used in other software. It has no dependencies and is being released under the BSD three-part license.
While I created these tools to scratch my own itch, I hope other people will find them useful. I hope in particular that researchers with limited access to powerful hardware can use this to break into genomics and statistical genetics. I would love to get user feedback, especially if there are any problems.